HQ Team
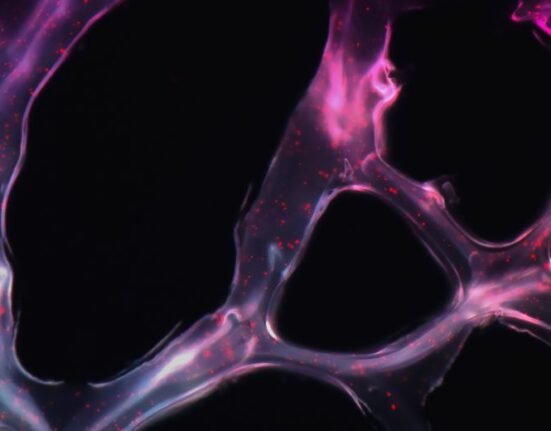
January 19, 2025: A study led by researchers from the Broad Institute, Harvard Medical School, and McLean Hospital has shed light on the genetic mechanisms underlying Huntington’s disease (HD). This fatal neurodegenerative disorder causes nerve cells in the brain to die.
The research revealed how somatic DNA mutation in the Huntington (HTT) gene contributes to the delayed onset of Huntington’s symptoms in patients.
Understanding Huntington’s disease
Researchers have known for decades that Huntington’s disease is caused by an inherited mutation in the HTT gene. But they didn’t know how the mutation increased beyond a threshold, and why only some brain cells die and not others.
Huntington’s disease is characterized by an abnormal expansion of a CAG repeat sequence in the HTT gene. While healthy individuals typically possess 15-35 CAG repeats, those with HD have over 40 repeats, often exceeding 150 as the disease progresses. This study provides critical insights into why symptoms of HD usually manifest in midlife despite individuals being born with the mutation.
Key findings
The research team utilized advanced techniques such as single-cell RNA sequencing to analyze brain tissue from 53 individuals diagnosed with HD. They discovered that the CAG repeat sequence expands slowly over decades within specific neurons, particularly in the striatum, before rapidly lengthening and leading to cell death. Notably, once the repeat count surpasses 80, the rate of expansion accelerates significantly.
The study highlights a process termed “somatic expansion,” where CAG repeats increase in length within neurons over time. This gradual accumulation eventually results in neurodegeneration when the repeat count exceeds critical thresholds.
Clinical trials concentrated on lowering the levels of the protein produced by the mutated Huntington’s gene have been unsuccessful. The new findings suggest that’s because few cells have the toxic version of the protein at any given time.
Slowing or stopping the expansion of DNA repeats may be a better way to target the disease, researchers said.
The current therapies focusing solely on reducing HTT expression may not be sufficient. Instead, targeting the mechanisms that slow down somatic expansion could provide new avenues for delaying or even preventing symptom onset in individuals at risk.
Future directions
The researchers emphasize that understanding the trajectory of HD pathology opens up multiple windows for treatment strategies. By slowing down the somatic expansion process, it may be possible to postpone the onset of symptoms significantly. This approach not only benefits those at risk but could also help individuals already exhibiting early signs of the disease.
Dr. Sabina Berretta from Harvard Medical School stated, “The point of our work—what we all do—is relieving suffering caused by disease,” underscoring the study’s potential impact on future treatments for Huntington’s and other similar disorders involving DNA repeat expansions.
The study is published in the journal Cell.